雷替曲塞
物理性質
性狀:淡黃色粉末
熔點(oC):180~184
溶解性:溶于水。
性質與穩定性
1.胸苷酸合成酶抑制劑。用于治療結腸、直腸癌。
2.對結腸癌細胞系的抑制作用較強
3.本品為胸苷酸合酶抑制劑.此藥的功效與5-氟尿嘧啶加亞葉酸的功效(總有效率)相似,但可使患者存活時間延長,改善生命質量等.臨床用于發展期直腸癌和結腸癌的治療.
合成方法
1. 2,6-二甲基-4(3H)-喹唑啉酮(I)用叔戊酰氧基甲基氯進行N烷化卮再用N-溴代丁二酰亞胺溴化,得到6-(溴甲基)-2-甲基-3-[(叔戊酰氧基)甲基]-4(3H)-喹唑啉酮(Ⅲ).(m)和N-[5-甲氨基噻吩-2-羰基]-L-谷氨酸二乙酯(IV)縮合,生成物V和氫氧化鈉的乙醇水溶液在室溫下攪拌水解,并同時脫保護基, 然后酸化得到產物.
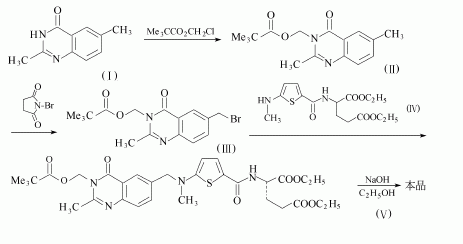
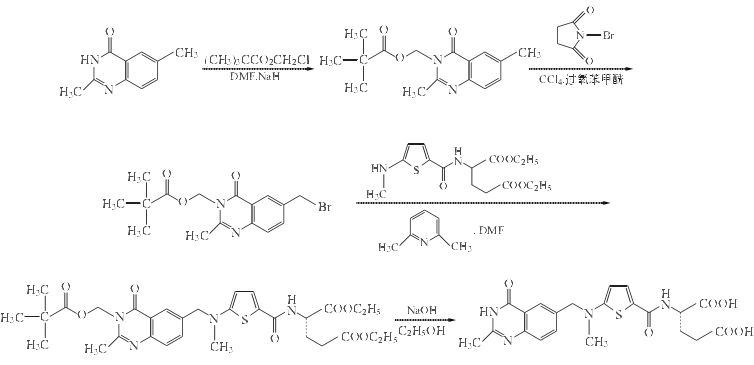
2.3,4-二氫-2,6-二甲基-3-新戊酰氧基甲基/喹唑啉-4-酮的制備
在反應瓶中加入3,4-二氫-2,6-二甲基喹唑啉-4-酮(按文獻[9]方法制備或外購)3.0g和二甲基甲酰胺50ml,攪拌溶解,在室溫攪拌下向該溶液加入氫化鈉(NaH)1.08g,將混合物于室溫攪拌1h.再加入氯甲基新戊酸酯3.36g溶
于二甲基甲酰胺10ml的溶液,混合物于室溫攪拌反應18h.然后將反應液傾入飽和食鹽水200ml中,用乙醚(50ml×4)提取,合并乙醚層,無水NaSO4干燥.過濾,濾液蒸發至干,剩余物經硅膠柱色譜純化[洗脫劑:二氯甲烷/乙酸乙酯(體積比9:1)].經后處理,產物再用石油醚(bp60~80 ºC)結晶,得3,4-二氫-2,6-二甲基-3-新戊酰氧基甲基/喹唑啉-4-酮0.92g,mp98~100 ºC.
3. 6-溴甲基-3,4-二氫-2-甲基-3-新戊酰氧基甲基喹唑啉-4-酮的制備
在反應瓶中加入上述化合物3,4-二氫-2,6-二甲基-3-新戊酰氧基甲基/喹唑啉-4-酮0.92g、四氯
化碳50ml、N-溴代琥珀酰亞胺0.624g和過氧苯甲酰0.025g(催化量)攪拌混合,緩慢升溫至回流,在回流下攪拌反應2h.冷卻至室溫將反應液通過裝填硅酸鎂25h的柱色譜,用四氯化碳洗脫,洗脫液減壓下蒸發至干,得固體剩余物6-溴甲基-3,4-二氫-2-甲基-3-新戊酰氧基甲基喹唑啉-4-酮6-溴甲基-3,4-二氫-2-甲基-3-新戊酰氧基甲基喹唑啉-4-酮1.6g,mp144~145 ºC.
4.N-[5-(甲基氨基)-2-噻吩甲酰基]-L-谷氨酸二乙酯的制備
在反應瓶中加入2-噻吩羧酸300g(2.34mol)、叔丁醇2500ml,攪拌溶解.在保持溫度恒定在20~25C下滴加三乙胺325ml(2.34mol),滴畢,再加入二苯基磷酰疊氮化物(DPPA)517ml(2.4mol). 將混合物攪拌回流12h.冷卻,黃色反應液傾倒入冰水7000ml中, 產生白色沉淀,過濾#濾餅用水洗凈,于35 ºC下真空干燥得2-[N-(叔丁氧羰基)氨基],噻吩431g,收率92%,mp147~148 ºC.
在滴液漏斗中加入2-[N-(叔丁氧羰基)氨基]噻吩100g(0.502mol)溶于DMF300ml溶液,將其滴入懸浮于DMF300ml中的NaH23.1g(分散于油中,55%含量,0.53mol) 的懸浮液中(滴加過程中溫度不超過5 ºC,并用氬氣保護,用冰浴冷卻).滴畢,在0~5 ºC繼續攪拌30min,再在攪拌下和15 ºC以下滴加甲基碘3.14g(0.50mol).滴畢,再在20 ºC連續攪拌16h.加入水200ml終止反應,用乙醚(1500ml×4)提取,合并乙醚層,用水洗和鹽水
洗,干燥,過濾,濾液濃縮蒸除溶劑,得2-[N-(叔丁氧羰基)-N-甲基氨基]噻吩的黃色油狀物123g,將其用硅膠柱色譜純化[洗脫劑為己烷/二氯甲烷(體積比1:1)]得精制的2-[N-(叔丁氧羰基)-N-甲基氨基]噻吩油狀物83.3g, 收率78%.
反應瓶中加入二異丙胺30ml0.21mol)和THF200ml攪拌溶解,在氬氣保護下,于-78 ºC滴加1.6mol/L正丁基鋰的己烷溶液119ml,滴加過程中應保持溫度低于-50 ºC.滴畢,于-78 ºC下攪拌反應30min.然后在低于-60 ºC下滴加2-[N-(叔丁氧羰基)-N-甲基氨基]噻吩40.8g(0.19mol)溶于THF200ml的溶液,滴畢,在-78 ºC繼續攪拌反應30min.然后在防潮情況下小心地少量分批加入粉碎的固體CO2200g(4.54mol)( 保持溫度低于-50 ºC).將反應混合物慢慢升溫至20 ºC.在該溫度下攪拌反應過夜.將反應液倒入水2L中,加入粉狀檸檬酸調至pH5,過濾出白色沉淀固體,水洗,真空干燥得5-[N-(叔丁氧基羰基)-N-甲基氨基]-2-噻吩羧酸(A)39.8g,收率81%,mp210~211 ºC (分解)
在反應瓶中加入上步產物39g(0.152mol)和DMF60ml溶于CH2Cl2 300ml,溶液,攪拌,于30min.內向該混合物加入草酰氯22.9g(0.27mol), 加畢,繼續攪拌30min后,將反應液低于30ºC下蒸發至干,得粗制的酰基氯黏性固體在真空下(0.1mmHg)干燥1h.
在反應瓶中加入谷氨酸二乙酯鹽酸鹽36.15g(0.152mol)和CH2Cl2 260ml ,攪拌溶解,在氬氣保護下加入三乙胺34.0g(0.24mol), 在冰浴冷卻下,冷至10ºC,再加入上述制備的粗制酰基氯溶于CH2Cl2 200ml溶液. 加料時保持溫度低于15 ºC ,加畢,于同溫度下攪拌反應過夜.得到暗綠色混合物,用水(200ml×2)洗滌,水層(水洗液)用CH2Cl2 400ml提取,合并有機層,干燥,濾去干燥劑的濾液蒸除溶劑得黑色剩余物,將其通過硅膠柱色譜純化[洗脫劑為0~5%(體積分數)梯度的乙酸乙酯的CH2Cl2溶液],得粗制谷氨酸衍生物74g.在反應瓶中加入谷氨酸衍生物74g和CF3COOH3 50ml,控溫20~25 ºC下攪拌反應16h后將CF3COOH3蒸除#剩余物加入NaHCO3水溶液1L, 用CH2Cl2(500ml×3) 提取,合并CH2Cl2層,用無水Na2SO4干燥,過濾,濾液濃縮得油狀物,將其油狀物經硅膠柱色譜純化[洗脫劑為10%(體積分數)乙酸乙酯的CH2Cl2溶液],得N-[5-(甲基氨基)-2-噻吩甲酰基]-L-谷氨酸二乙酯33.35g(為黏稠棕色油狀物),收率58%.
5.N-[[5-[[(1,4-二氫-2-甲基-3-新戊酰氧甲基-4-氧-6-喹唑啉基)甲基]甲氨基]-2-噻吩基]L-谷氨酸二乙酯的制備
在反應瓶中加入N-[5-(甲基氨基)-2-噻吩甲酰基]-L-谷氨酸二乙酯1.5g(4.4mmol) 、6-溴甲基-3,4-二氫-2-甲基-3-新戊酰氧基甲基喹唑啉-4-酮1.71g(4.7mmol) 、2,6-二甲基吡啶0.47g(510μl,4.4mol)和DMF26ml,攪拌溶解.該混合物于60 ºC和氬氣保護下攪拌反應18h.冷卻#混合物在低于40ºC下蒸發至干,余物經硅膠柱色譜純化(脫劑:1%甲醇的乙酸乙酯溶液),得N-[[5-[[(1,4-二氫-2-甲基-3-新戊酰氧甲基-4-氧-6-喹唑啉基)甲基]甲氨基]-2-噻吩基]L-谷氨酸二乙酯340mg, 收率15%
6. N-[[5-[[(1,4-二氫-2-甲基-4-氧-6-喹唑啉基)甲基]甲氨基]-2-噻吩基]羰基]-L-谷氨酸(蘭替特噻)的合成
在反應瓶中加入N-[[5-[[(1,4-二氫-2-甲基-3-新戊酰氧甲基-4-氧-6-喹唑啉基)甲基]甲氨基]-2-噻吩基]L-谷氨酸二乙酯340mg(0.66mmol)、加入5ml和1mol/LNaOH水溶液6.6ml(6.6mmol),在氬氣保護下攪拌反應2h.將反應液過濾,液用2mol/L鹽酸調至Ph3.0,生沉淀,沉淀過濾,濾餅用水洗至無鹵離子存在.潮濕產物經干燥,得129mg, 收率41%,mp180~184 ºC.
分子結構數據
1、 摩爾折射率:117.35
2、 摩爾體積(m3/mol):306.1
3、 等張比容(90.2K):306.1
4、 表面張力(dyne/cm):63.0
5、 極化率(10-24cm3):46.52
安全信息
危險運輸編碼: UN 2811 6.1/PG 3
危險品標志: 有毒
安全標識:S45 S53
危險標識:R25 R61







